-
Rigid Bis(tetrathiafulvalenes) Doubly Bridged by Phosphino Groups and Derivatives: Synthesis and Intramolecular Mixed Valence State

I. Danila, F. Biaso, H. Sidorenkova, M. Geoffroy, M. Fourmigué, E. Levillain and N. Avarvari
Organometallics, 28 (13) (2009), p3691-3699


DOI:10.1021/om900107y | unige:3551 | Abstract | Article HTML | Article PDF
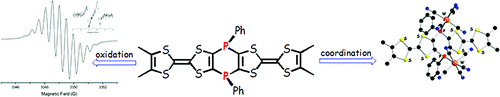
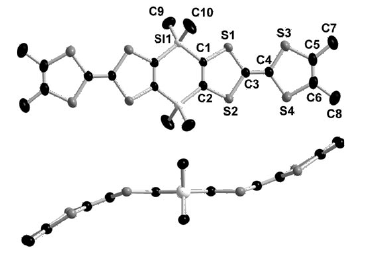
The synthesis and structural characterization of the λ5-bis(phosphine sulfide) and the bimetalliccomplexes bis[phosphino-M(CO)5] (M = Mo, W) of the 3,4-dimethyltetrathiafulvalene (ortho-DMTTF)-based rigid dimer (PPh)2(o-DMTTF)2, containing a central 1,4-dihydro-1,4-diphosphi-nine ring, are described. Single-crystal X-ray analyses have been performed for the trans isomers(PhPX)2(o-DMTTF)2 (X = S, Mo(CO)5, and W(CO)5) and for the cis isomer [PhPW(CO)5]2-(o-DMTTF)2. Planar or slightly folded boat-type conformations are observed for the central six-membered ring, together with different packings characterized by short intermolecular S · · · Scontacts. The optical signature of the oxidized species in the case of the free ligand (PPh)2-(o-DMTTF)2 has been evidenced by UV-vis spectroelectrochemistry measurements. SolutionEPR measurements on the radical cation species of (PPh)2(o-DMTTF)2 definitely assess the fulldelocalization of the unpaired electron over both electroactive TTF units, with an associatedcoupling of 0.48 G with 12 equivalent protons. The EPR signal of the dication proves the radicalnature of this species, in favor of a triplet ground state. The radical cation of the cis-[PhPW(CO)5]2-(o-DMTTF)2 isomer was also investigated by EPR, for which the observed hyperfine structuredemonstrates the extended delocalization of the electron, together with a larger coupling constantwith the phosphorus nuclei. DFT calculations for the radical cation of (PPh)2(o-DMTTF)2 afford aboat-type conformation for the central ring and a SOMO consistent with a full delocalization of theelectron over both TTF units. Moreover, the calculations indicate that in the case of the dication of(PPh)2(o-DMTTF)2 the triplet state is more stable by 11.7 kcal mol-1 than the singlet state.